Editora: Lisiane Martins.
Colaboradores: Amanda Lewandowski da Silva, Giovani Meneguzzi de Carvalho, João Francisco Petry, Mariana Ribeiro e Pâmella Kreling.
Conceito de Farmacologia[]
A farmacologia é a ciência que estuda os efeitos das substâncias químicas sobre a função dos sistemas biológicos, ou seja, o que ocorre com o medicamento no nosso organismo desde o momento em que é administrado. Isso também depende da via pela qual é administrado, como será comentado em “Vias de Administração de Fármacos". A farmacologia envolve a absorção, distribuição, estudo do efeito que o fármaco vai gerar no organismo e a maneira pela qual irá ser metabolizado a fim de que seja eliminado. Cada vez que uma classe de medicamento for citada, esses efeitos serão avaliados. Dessa maneira, para organizar o estudo da farmacologia, tem-se farmacocinética e farmacodinâmica.
Farmacocinética[]
A farmacocinética estuda a absorção, distribuição, metabolização e eliminação dos medicamentos, enquanto a farmacodinâmica irá estudar o mecanismo de ação dos medicamentos (onde irá se ligar para gerar efeito no organismo, seja a um receptor, em alguma célula, inibindo uma enzima, ou ativando um receptor, por exemplo). Isso é denominado como um mecanismo de ação, ou seja, o que o medicamento irá fazer para gerar um efeito. A finalidade dos medicamentos é atingir a homeostasia. Eles podem estar disponíveis em forma de pastilha, xarope, comprimido, supositório, suspensão, cápsula, drágea, ampola, etc.
Absorção: transferência do fármaco desde seu local de aplicação até a corrente circulatória.
Biotransformação: modifica a estrutura do fármaco, inativando-o (na maioria das vezes) e facilitando sua eliminação.
Excreção: saída do fármaco do organismo.
Antes da discussão farmacocinética, em 1970, os esquemas de administração eram decididos através de observação clínica, por ensaio X erro, até se conseguir doses eficazes (resposta terapêutica) ou excessivas (reações tóxicas).
Intervalos entre as doses: calculados em função do tempo de eliminação do fármaco.
A prescrição médica deve conter:
- Dose
- Via de administração
- Intervalo entre as doses
Isso é muito importante, principalmente quando a concentração terapêutica for muito próxima da tóxica (antiarrítmicos, anticonvulsivantes, etc).
Quanto maior a distância entre a concentração terapêutica e a tóxica, menos críticos são os parâmetros farmacocinéticos.
|
Concentração em sítio-alvo |
Efeitos |
|
Excessiva |
Tóxicos |
|
Máxima permitida |
Potencialmente tóxicos |
|
Ótima |
Terapêuticos |
|
Limiar |
Parcialmente eficazes |
|
Insuficiente |
Ausentes |
Propriedades dos Fármacos[]
Para que ocorra a absorção, distribuição, metabolismo e eliminação desses medicamentos, eles devem ser transportados através das membranas celulares. Para que esse transporte ocorra, eles precisam possuir algumas características/propriedades que vão facilitar ou reduzir a capacidade de serem transportados:
- Peso molecular: porque quando menor o PM, mais fácil será o seu transporte através das membranas;
- Configuração da substância (configuração estrutural): essa confirguração irá ou não possibilitar transporte, através da ligação a receptores de membrana e a proteínas transportadoras para que ele possa ser transportado;
- Grau de ionização (dissociação): pois o fármaco ionizado não sofre transporte através da membrana. É o não-ionizado que sofre esse transporte. Assim, ter 90% de fármaco não-ionizado e 10% ionizado é melhor do que ter 50% não-ionizado e 50% ionizado;
- Lipossolubilidade: Quanto mais lipossolúvel, mais fácil será o transporte através das membranas;
- Ligação às proteínas: o que realmente auxilia no transporte dos compostos polares são as proteínas transportadoras. Isso inclui tanto as proteínas plasmáticas quanto as teciduais.
Os fármacos possuem uma fração ligada à proteína e uma fração livre. No plasma, a albumina é a principal proteína que se liga aos medicamentos. A fração ligada não é ativa. Isso porque quando um fármaco se liga às proteínas, ele forma um complexo grande (e as proteínas já possuem o peso molecular alto). Portanto, se ligada ao fármaco, forma um complexo (fármaco+proteína) que não vai sofrer transporte pela membrana.
A fração livre (que não se ligou à proteína) terá menor PM, sendo chamada de fração ativa. A fração livre é a ativa, e é a que interessa, pois sofre transporte através das membranas. Assim, se estiver ligado a proteínas, o fármaco não será transportado. A fração livre está em menor proporção (o que depende de cada medicamento) e vai chegar ao seu local de ação para proporcionar o efeito.
As proteínas teciduais também estão incluídas porque se o medicamento chegar num tecido e se ligar a uma proteína (ficar complexado) independente de ser ou não o local dele, ele não poderá ser transportado para outros locais. Esse fármaco poderá ficar ligado num local que é reservatório e depois, aos poucos, pode retornar para a corrente sanguínea. Mas enquanto estiver ligado, ele não estará atuando onde deveria estar. Então há uma dependência tanto da ligação a proteínas plasmáticas, quanto às teciduais.
Membranas Celulares[]

{kind=link}
Membrana ceular - Fonte: Daniela Delwing de Lima, 2013 (apresentação ppt feita em sala.
A membrana das células é composta de uma bicamada lipídica, e possui o fluido extracelular. Essa bicamada é composta principalmente por fosfolipídeos, que são moléculas anfipáticas, pois têm uma fração polar e outra apolar. A fração polar corresponde aos grupamentos fosfato, que por serem polares se orientam para o meio extracelular (também é polar) e para o citoplasma da célula (que também é polar). Já a fração de ácido graxo é formada por hidrocarbonetos (carbono e hidrogênio) e se orienta para o centro dessa bicamada lipídica. Essas moléculas de lipídeo também podem se ligar a moléculas de colesterol e regular a fluidez da membrana, a flexibilidade, a organização e torná-la impermeável a moléculas polares. A bicamada, por ser apolar, é impermeável a moléculas polares (e permeável a apolares). Dessa maneira, o fármaco lipossolúvel é transportado facilmente, porque é solúvel em lipídeos e sofre difusão fácil através da membrana. Já os fármacos polares não conseguem ser transportados pela bicamada lipídica, por isso nessa membrana existem as proteínas. Elas podem atuar como proteínas integrais (transmembrana) que atravessam toda a bicamada, ou como proteínas periféricas. As proteínas periféricas estão ligadas a proteínas integrais ou à superfície externa da membrana.
Tais proteínas, para conseguirem atravessar a membrana, são compostas na parte apolar por aminoácidos apolares, o que possibilita que atravessem a bicamada. Na parte voltada para o meio extracelular e o citoplasma, contêm aminoácidos polares (o que possibilita que também possa se orientar para o citoplasma e para o meio extracelular). A diferença entre as proteínas integrais e as periféricas é que as periféricas geralmente vão atuar como enzimas e também são alvo da ação de medicamentos. Isso porque alguns medicamentos têm seu efeito voltado para atuar sobre as enzimas (estimular ou inibir).
Transporte Passivo[]
As proteínas integrais vão atuar nos canais iônicos e nas proteínas transportadoras. Esses canais iônicos e proteínas transportadoras possuem receptores para ligação de substâncias (tanto do organismo quanto de fármacos). Os canais iônicos (ex: aquaporinas) não funcionam muito para transporte de medicamentos, porque possuem diâmetro pequeno. O que realmente auxilia no transporte de medicamentos polares são as proteínas transportadoras.
Além disso, essas membranas são consideradas relativamente permeáveis à água. Isso ocorre dependendo dos mecanismos de difusão (a favor do gradiente de concentração, de onde tem mais para onde há menos) e também dos gradientes formados através da membrana (gradientes transmembrana) que envolvem o gradiente osmótico e o gradiente hidrostático. O gradiente osmóstico se refere a [ ] de solutos, enquanto o hidrostático à [ ] de líquidos. Então, se houver uma membrana e uma alta [ ] de soluto de um lado, isso gerará um gradiente osmótico transmembrana. Assim, a água sofrerá transporte para o lado com mais soluto (efeito osmótico). Quando a água sofre esse efeito osmótico em função da [ ] de soluto, tem-se um gradiente hidrostático (aumento de pressão hidrostática) que favorecerá o fluxo de líquidos através das membranas. Esse processo é chamado de mecanismo por convecção ou dragagem pelo solvente.
O transporte da água também possibilita que a água leve com ela algumas moléculas de soluto, quando sofre o efeito osmótico, inclusive fármacos. No entanto, um fármaco pode estar presente ligado à proteína ou livre. Assim, se estiver ligado à proteína não pode sofrer esse efeito osmótico, pois o complexo será de alto peso molecular e a água não irá conseguir transportá-lo. Somente o fármaco livre pode ser transportado juntamente com a água, se o seu peso molecular for também adequado. O limite para transporte desse efeito é de até 200 daltons.
Sempre é mantida uma proporção dentro de diferentes compartimentos e as frações ligadas/desligadas de proteínas, a fim de manter o equilíbrio. Quando o fármaco livre é transportado para o seu tecido de ação, diminui a fração do plasma, e outra fração ligada se desliga da proteína para também sofrer transporte (e assim sucessivamente). Isso tem um limite, para que devido a esse mecanismo de desligamento não haja somente fármacos livres no organismo (o que traria um efeito imensamente maior, e não é o que se espera). Esse transporte é transcelular (através da célula) e também paracelular, (entre as células) pelo espaço intercelular. O transporte entre as células é muito usado nas células endoteliais dos capilares, pois a união das células não é tão firme (junções oclusivas). Isso é importante para os fármacos que não forem lipofílicos, pois podem ter esse tipo de transporte e permitir a passagem de medicamento.

{kind=link}
Barreira Hematoencefálica - Fonte: http://2.bp.blogspot.com/-ReolbSy2iZA/UA6ZamNc7eI/AAAAAAAAAJE/aJL2wHOx8G4/s1600/Untitled.png
Todavia, esse tipo de transporte não ocorre em alguns locais do organismo, como por exemplo o SNC, pois as células endoteliais dos capilares cerebrais estão unidas por junções íntimas. Essa barreira pode ser hematoencefálica (com células endoteliais dos capilares cerebrais firmemente conectadas e sem frestas entre elas) e liquórica. As células gliais auxiliam essa comunicação e inviabilizam a passagem de medicamentos pelo meio paracelular. Assim, não ocorre esse tipo de transporte (o que limita o transporte paracelular é o fluxo sanguíneo, pois quanto maior esse fluxo, mais fácil o transporte). O transporte paracelular dos fármacos polares costuma ser mais rápido, pois não precisa atravessar membranas. Numa meningite, no entanto, os fármacos podem ter acesso ao SNC devido ao aumento da permeabilidade que ocorre.
Transporte dos medicamentos:

{kind=link}
Transporte através da Membrana - Fonte: http://resumosdosegunda.files.wordpress.com/2011/08/1.jpg
O transporte é algo extremamente importante, pois é disso que os medicamentos dependerão para atuar. O principal mecanismo de transporte do fármaco através das membranas é o transporte passivo, sendo que a maior parte dos medicamentos são transportadas dessa forma. Então, se é por transporte passivo, este ocorre por difusão, dependendo de um gradiente de concentração (a favor do mesmo), que também dependerá da lipossolubilidade do medicamento, pois vai estar associada a sua solubilidade na bicamada lipídica. Quando mais lipossolúvel, mais fácil a difusão. A difusão depende também, portanto, do coeficiente de partição hidrolipídica. Esse coeficiente nos diz quanto de um medicamento é solúvel em água e quanto é solúvel em lipídeo.
Para que sofra difusão pela bicamada, é preciso que seja mais solúvel em lipídio, dependendo de vários coeficientes de partição de lipídio. Segundo a professora, o livro Goodman está errado, pois diz que depende de um ALTO coeficiente de partição hidrolipídica, mas segundo ela, depende de um baixo coeficiente ou de um alto coeficiente de partição óleo-água. Assim, se o coeficiente de partição for baixo, quer dizer que o medicamento é pouco solúvel em água e bastante solúvel em lipídeo. Quando mais baixo for, mais fácil será o transporte pela membrana. Além disso, quanto maior a área de membrana exposta, mais fácil e rápido será o processo de difusão. Após atingir o estado de equilíbrio, a [ ] do fármaco será a mesma dos dois lados da membrana, pois a difusão ocorre até que se estabeleça equilíbrio. Isso só ocorre com o fármaco livre (visto que o ligado não sofre difusão).
Compostos Iônicos
No caso de compostos iônicos, compreendem os fármacos que são ácidos fracos e bases fracas. Para esses fármacos, o estado de equilíbrio vai depender:
do gradiente de concentração do medicamento;
do pKa do fármaco e da diferença de pH transmembrana (entre os dois lados).
pKa: É o pH no qual a metade do fármaco (ácido ou base fraca) está em sua forma ionizada. Em outras palavras, o pKa é uma constante de dissociação, que diz que em um determinado pH, se terá 50% do fármaco ionizado e 50% do fármaco não-ionizado. Ex: pKa = 5. Dessa forma, no pH 5, 50% do medicamento estará ionizado, e 50% não ionizado.
Fármacos


{kind=link}
Absorção e eliminação - Fonte: Daniela Delwing de Lima, 2013 (apresentação ppt feita em sala)
Um grande número de fármacos são ácidos fracos e bases fracas e precisam estar dessa forma, a fim de que não sejam totalmente ionizados, pois não seriam absorvidos. Ou então, eles podem estar na forma não-ionizada, que é lipossolúvel e sofre transporte através das membranas. Com base nisso, a figura mostra como vai se comportar um ácido que é fraco e possui pH de 3,5 e uma base fraca com pH de 8,6. Tem-se três compartimentos (suco gástrico, plasma e urina – líquido no lúmen tubular). A barra mais escura representa a fração não-ionizada, e barra mais clara diz respeito a fração ionizada do medicamento. No ácido fraco, a fração não-ionizada está em equilíbrio nos diferentes compartimentos (não tem interferência na difusão). A fração livre (à medida que o pH foi aumentando), aumentou também a fração ionizada desses ácidos fracos – aumentou a liberação de prótons no meio e o fármaco foi se ionizando. A maior fração livre tem pH mais alto, inclusive. Já em relação a base fraca,a medida que o pH se torna mais ácido, mais ionização a base terá, fazendo com que se acumule mais num meio mais ácido (coeficiente de partição de pH).
Equação de Henderson-Hasselbalch
pH = pKa + log([A-]/[HA])
Conforme dito anteriormente, o fármaco não ionizado passa pela membrana, e o ionizado não o faz. Ele se ioniza porque vai mudando o pH, que se torna muito diferente do pKa dele. Na urina, que é alcalina, mais ionização ela irá receber. Assim, libera prótons para manter o equilíbrio do meio. Quando o pH vai caindo, começa a liberar hidroxila.
Um fármaco ácido fraco sempre vai se acumular mais num meio mais alcalino, pois lá estará mais ionizada (forma que não sofre transporte). Um fármaco que é uma base fraca, por outro lado, se acumulará mais num meio mais ácido, pois nesse meio estará mais ionizado e não sofrerá difusão. Esses conceitos interferem tanto na absorção quanto na eliminação.
Ex.: urina (pH 8): uma grande quantidade estará no lúmen tubular e não poderá ser reabsorvido (será eliminado). Somente o que ficou de fármaco não ionizado poderá ser reabsorvido e voltar para o organismo. Isso é importante na intoxicação: a alcalinização da urina, depois de intoxicação por ácido acetilsalicílico favorecerá sua eliminação do organismo.
O fármaco que é um acido fraco é mais facilmente absorvido no estômago (está mais não-ionizado), e um fármaco que é base fraca é mais facilmente absorvido no intestino, devido ao meio ser mais alcalino. Dessa maneira, a absorção é favorecida quando o meio é mais parecido com o tipo do fármaco. Assim, para um ácido fraco ser absorvido no intestino, irá levar mais tempo. Não quer dizer que não será absorvido (pois a forma não-ionizada precisa se manter em equilíbrio). Então sofrerá difusão e diminuirá gradativamente o conteúdo do estômago, e uma fração ionizada terá que se tornar não-ionizada para dar continuidade ao processo. Isso, contudo, faz com que o medicamento seja absorvido mais lentamente. Assim, perde-se medicamento, pois se o medicamento for metabolizado por enzimas no intestino, essa degradação resultará em perda (mas mesmo assim será absorvido).
Sintetizando os fatores que influenciam os transportes:
§ Permeabilidade seletiva (Constituintes de membrana, sua inter-relação, polaridade, diâmetro dos poros);
§ Velocidade de transporte (espessura, área permeável da membrana, tamanho e forma molecular do fármaco).
Meio ácido:
o Bases fracas
o Forma ionizada
o Polar
o Mais hidrossolúvel
o Maior dificuldade para transpor a membrana
Meio alcalino:
o Processos inversos
o Formas não-ionizadas
o pH pode ser modificado (mediante adição de ácidos ou bases, com a finalidade terapêutica de facilitar ou dificultar os transportes através de membranas).
Transporte (Carreadores)[]
Difusão Passiva[]

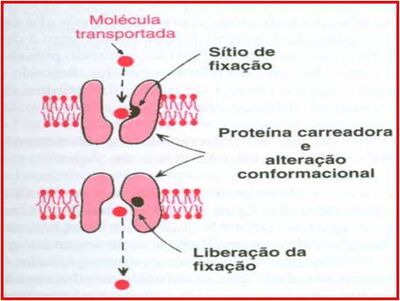
{kind=link}
Difusão - Fonte: Daniela Delwing de Lima, 2013 (apresentação ppt feita em sala)
Difusão facilitada: a favor do gradiente de [ ] e com velocidade superior à da difusão simples. Não utiliza energia por hidrólise de ATP (pois é a favor do gradiente), e depende de uma proteína transportadora (transmembrana) que possui um sítio de ligação. Assim, a substância a ser transportada vai se ligar ao sítio de ligação, precisando ter conformação semelhante a este. O transporte é importante para substâncias maiores (polares) que dependem de um carreador. Após a ligação, ocorre alteração conformacional, para que do lado externo se abra para o interno, a fim de que ocorra ligação da substância.
Ex: transportador de glicose – GLUT 4 – é dependente de insulina e se localiza na membrana da célula muscular. Ele é uma proteína transmembrana (integral) e sua síntese é estimulada pela insulina. Quanto mais insulina, mais glicose para o sangre. Então o pâncreas libera insulina, que aumenta a expressão/síntese de GLUT 4, o que possibilita a captação de glicose pela célula muscular (por processo de difusão facilitada), para ser utilizada como fonte de energia.
Os fármacos não transportados por difusão (minoria):
Transporte ativo[]
Contra gradiente de concentração (energia por hidrólise de ATP ou de outras ligações altamente energéticas):
- Pode ser dividido em primário e secundário:
Primário []
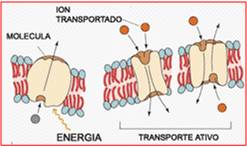
{kind=link}
Transporte ativo secundário - Fonte:
É contra gradiente de [ ] e utiliza energia proveniente da hidrólise do ATP ou outro nucleotídeo. Se caracteriza por saturabilidade (é saturável, tem sítios de ligação que quando estão todos preenchidos, outra substância não pode se ligar), seletivo (no sítio de ligação do sódio não há como o potássio se ligar, por exemplo) e pode ter competição de fármacos (inibição competitiva). Ex: Bomba de sódio-potássio-ATPase.
Ex: Digoxina (glicosídio digitálico), que compete com o potássio pelo sítio de ligação – impede que ele se ligue – aumentando a força de contração do músculo cardíaco, sendo usada em casos de insuficiência cardíaca.
Obs.: Bomba de sódio-potássio-magnésio-ATPase se referem à mesma bmba, pois o magnésio pode estar na bomba já que ele é um co-fator (a bomba precisa de magnésio para funcionar). A bomba está presente em todas as células e é responsável por “arrumar” os gradientes de concentração (manter a concentração de sódio correta no meio extracelular, e de potássio no meio intracelular) no intuito de manter o repouso da célula. O sódio é um cátion, presente em alta concentração no meio extracelular. Já o potássio é cátion presente em alta concentração no meio intracelular. A bomba vai contra gradiente: bombeia sódio do meio intracelular para o meio extracelular, e potássio do meio extracelular para o meio intracelular. Os potenciais ocorrem com entrada de sódio e saída de potássio, então a bomba irá reorganizar o processo. Existem três sítios de ligação para o sódio e dois sítios de ligação para o potássio. O sódio sempre se liga primeiro. Para sair da célula, a ATPase hidrolisa o ATP, que libera fosfato no processo. O fosfato se liga a bomba, o que significa que ela foi fosforilada e vai sofrer alteração conformacional para que os três sódios possam ser liberados para o meio extracelular. Nesse momento, ocorre liberação de sódio e os dois potássios se ligam à bomba, e o grupamento fosfato é liberado (desfosforilação). Irá sofrer então alteração conformacional para que os dois potássios possam ser transportados para o meio intracelular.
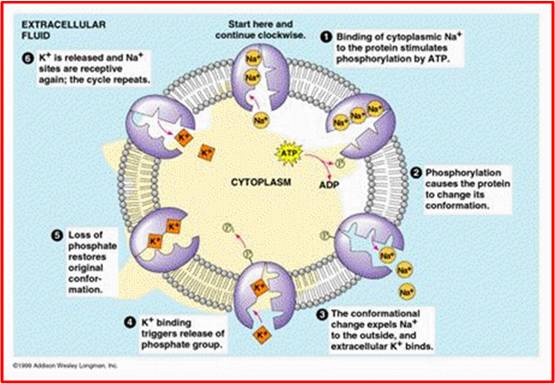
{kind=link}
Transporte ativo - Fonte: Daniela Delwing de Lima, 2013 (apresentação ppt feita em sala)
Secundário
O outro tipo de transporte ativo. Nele, a energia é gerada por um gradiente de concentração transmembrana (uma diferença de [ ] entre os dois lados da membrana). O gradiente é gerado pelos íons, principalmente pelos íons sódio, potássio e cloreto (os três são capazes de gerar energia, pois são os íons em maior concentração ou no meio extra ou no intracelular (Por exemplo o cloreto: é mais abundante no meio extracelular, também podendo gerar gradiente de concentração).
Esse transporte secundário vai se dividir em contra-transporte ou antiporte (que possuem o mesmo significado) e co-transporte ou simporte (também são sinônimos).
Co-transporte (transporte para a mesma direção) – proteína integral que possui dois sítios de ligação orientados para o mesmo meio (extracelular ou intracelular). Se o transporte for para o meio intracelular, quem fornece energia é o potássio.
Ex: sódio-glicose – o sódio se liga a um sítio e a glicose a outro. Quando os dois se ligarem, a proteína sofre alteração conformacional e o sódio é transportado para o meio intracelular (por difusão, a favor do gradiente) fornecendo energia para que a glicose também seja transportada para o mesmo meio. Assim, o transporte da glicose utilizou energia proveniente do gradiente de concentração do sódio. Se o sódio não tivesse se ligado, a glicose não entraria na célula. Isso ocorre nas células epiteliais do trato gastrointestinal e também nos túbulos renais. O sódio pode retornar para o meio extracelular através de uma ATPase.
Contra-transporte: a diferença está nos sítios de ligação. Também é uma proteína transmembrana, mas um sítio de ligação está voltado para o meio extracelular e o outro para o meio intracelular.
Ex: sódio-cálcio. O cálcio se liga ao sítio voltado para o meio intracelular. A partir do momento em que os dois se ligaram, a proteína sofre alteração conformacional e o sódio entra por difusão na célula, fornecendo energia para que o cálcio saia da mesma (contra gradiente de concentração, visto que está indo para um local de maior concentração).
Transportadores de Eliminação
Além desses transportadores, existem outros, que são os transportadores de eliminação. É o caso da glicoproteína P, que auxilia na eliminação de medicamentos (tanto renal quanto a retirada do medicamento de um local que o organismo não quer que ele esteja). Existe no SNC, nos astrócitos, na membrana dos canalículos biliares, nos túbulos renais, na placenta, etc. O SNC vai restringir que um fármaco permaneça no mesmo, e a placenta vai impedir que o fármaco seja transportado até o feto.
Figura Resumo

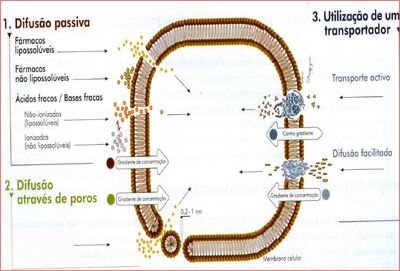
{kind=link}
Figura Resumo - Fonte: Daniela Delwing de Lima, 2013 (ppt utilizado em sala)
Essa figura é basicamente um resumo da aula, e foi requisitado pela professora que fosse preenchida.
Difusão passiva – fármaco lipossolúvel sofrerá facilmente difusão através da membrana. O não lipossolúvel não sofrerá facilmente essa difusão, e dependerá da difusão facilitada, por exemplo. Fármacos ácidos fracos e bases fracas estarão na forma não-ionizada (lipossolúvel e sofre difusão através das membranas) e ionizada (o contrário).
Através do poro: aquaporinas, por exemplo. Envolve canal iônico com pequeno diâmetro interno, não contribuindo muito para transporte de medicamento. O transporte de gases é mais efetivo (gás carbônico, oxigênio).
Difusão facilitada – fármacos polares, que têm proteína transmembrana que ajudará no transporte desse fármaco a favor do seu gradiente de concentração.
Transporte ativo – primário e secundário, contra o gradiente de [ ] – primário usa hidrólise de ATP e secundário usa energia da diferença de concentração iônica existente transmembrana de íons. Se divide em co-transporte (na mesma direção) e contra-transporte (direção oposta).
Absorção[]
Transferência de um fármaco do seu local de administração para o compartimento central e a amplitude com que isso ocorre. O compartimento central é a circulação sistêmica. A absorção do medicamento mostra a absorção do medicamento a partir da onde é administrado. Portanto, depende da via. Pode ser via oral ou intravenosa, por exemplo. Na intravenosa, já é administrado diretamente no compartimento central, não necessitando ser transferido. Se um fármaco é administrado via oral, será transportado, passará pelo estômago, intestino, sendo então absorvido e levado pela circulação-porta para a célula hepática e de lá atinge a circulação sistêmica.

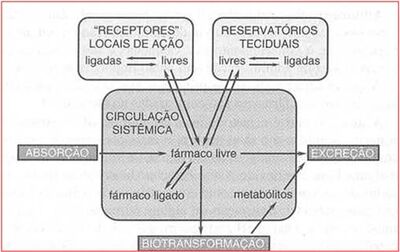
{kind=link}
Absorção - Daniela Delwing de Lima, 2013 (apresentação ppt feita em sala.
Portanto, existe um processo de transporte para que chegue à circulação sistêmica, o que demanda um tempo maior do que o fármaco que já foi administrado diretamente por via intravenosa. Uma fração do fármaco estará livre e outra se ligará as proteínas plasmáticas. A fração livre será transportada para seus locais de ação. No local de ação, a fração livre irá se ligar a receptores e exercer seu mecanismo de ação. A fração que se ligou, novamente através de uma constante de dissociação (o fármaco não ficara ligado todo o tempo), e o fármaco livre será novamente liberado e poderá retornar à circulação . Além do transporte para o local de ação, pode ser transportado para outros locais, em função do fluxo sanguíneo. Esses outros locais podem atuar como reservatórios teciduais, por afinidade. O tecido adiposo, por exemplo, é um reservatório de fármacos.
Como exemplo, os benzodiazepínicos são utilizados, pois o tecido adiposo é lipofílico e o remédio também. Assim, devido ao transporte, acaba se acumulando e se transformando em reservatório de benzodiazepínico. Uma vez livre, o medicamento pode retornar para a circulação sistêmica. Se o fármaco permanecer acumulado, pode gerar uma hepatotoxicidade (no caso do fígado, por exemplo), uma fibrose (no caso do pulmão), etc. Por isso é importante que o medicamento retorne à forma livre. Quando o fármaco retorna do local de depósito, volta à circulação sistêmica e é transportado novamente para outros locais (como por exemplo o fígado - ocorrência da maior parte do metabolismo), onde serão metabolizados para poderem ser eliminados. A principal via de eliminação é a via renal. No entanto, alguma quantidade é eliminada também pelas fezes (como os sequestradores de ácidos biliares).
Fármacos por via oral: a taxa de absorção para circulação sistêmica também vai depender da preparação do fármaco. A preparação sólida depende da dissolução da cápsula (da degradação do comprimido). Numa solução ou suspensão, a absorção é mais rápida que num comprimido.
Biodisponibilidade
Prescreve a percentagem na qual uma dose do fármaco chega no seu local de ação. Assim, se for um fármaco injetado pela via intravenosa, a biodisponibilidade é alta, pois chega rapidamente no local em que vai atuar. Por via oral, o medicamento pode iniciar a degradação já no estômago (devido ao ph ácido) que para alguns fármacos não é bom, por já dar início ao processo degradativo, de destruição do medicamento. No intestino, existem enzimas que degradam o fármaco e fazem o metabolismo do mesmo, mesmo antes de ser absorvido. Então, há perda de medicamento. Do intestino ele irá para a célula hepática, onde também há enzima que metaboliza e, novamente, há perda de fármaco. Após o fígado, o medicamento vai para a circulação sistêmica, e a biodisponibilidade desse fármaco é diminuída. Esse é o metabolismo de primeira passagem (que não pode ser chamado de metabolismo hepático, porque também ocorre no intestino). É assim denominado porque não havia a intenção dessa perda de medicamento, que sempre vai depender da via de administração. Se a capacidade do fígado de metabolizar ou de excretar (para a bile) for grande, isso levará a uma grande queda na biodisponibilidade do fármaco.
Referências
Links Externos