Editor: Giovani Meneguzzi de Carvalho
Colaboradores: Amanda Lewandowski da Silva, Lisiane Martins, João Francisco Petry, Mariana Ribeiro e Silva e Pâmella Kreling
CÁLCIO[]
Quanto a entrada na célula:[]
A concentração de cálcio no interior da célula vai depender de sua entrada a partir dos canais de cálcio na membrana plasmática, que são regulados pela proteína G. Sendo assim, se a proteína G se liga a estes canais, ela provoca a abertura e possibilita a entrada de Ca.
A concentração de cálcio intracelular também depende do potencial de membrana, devido as concentrações dos íons K e Ca que estão envolvidos nos processos de despolarização e repolarização.
Canais de Ca presentes na membrana do retículo endoplasmátimo, onde atua o IP-3 (trifosfato de inositol – do receptor tipo 2), liberam quantidades de Ca para o citoplasma a partir do RE quando o IP-3 se liga à estes canais.
Canais iônicos dependentes de voltagem em células excitáveis alteram as concentrações de Ca pela alteração conformacional do canal (antes estava no repouso e então foi ativado). Isto é o que acontece durante a despolarização da membrana. É importante notar que um canal iônico num estado inativado, primeiramente, tem que passar para o estado de repouso para então ser ativado novamente.
A quantidade de Ca no retículo sarcoplasmático também deve ser considerada, pois o RS é um local de armazenamento de íons Ca. Sendo assim, uma vez que seja estimulado o retículo sarcoplasmático promove a liberação destes íons.
Quanto a saída do Ca da célula:[]
Pode ser através do anti-porte, que é a troca sódio-cálcio. Entrada de Na enquando que o Ca sai da célula. Outro modo de remoção de Ca é através da bomba Cálcio ATPase, que transporta o Ca para o interior de organelas (retículo sarcoplasmático) a partir da hidrólise do ATP por transporte primário. O terceiro método de retirada do Ca intracelular é através da bomba SERCA, que transporta o Ca intracelular para dentro do retículo endoplasmático.
Funções do Cálcio: o Ca tem como função controlar o papel de exocitose das vesículas sinápticas, consequentemente regulação da liberação de neurotransmissores. Além disso, o cálcio tem importante papel na contração muscular, tanto na musculatura estriada quanto na lisa.
Regulação de Receptores:[]
Os receptores podem sofrer uma dessensibilização ou supersensibilidade.
Quanto a dessensibilização:[]
A dessensibilização também pode ser chamada de adaptação, hiporregulação ou refratariedade. Ocorre devido a estimulação prolongada da célula por um agonista. É o caso de um tratamento crônico. Devido ao uso contínuo do medicamento a resposta ao fármaco será atenuada. A causa é uma alteração no receptor que faz com que ele responda menos, pode ser por uma alteração conformacional, inacessiblidade temporária do receptor, redução de síntese de receptores ou por uma maior permanência ao estado inativado.
Uma das alternativas é trocar a classe do medicamento. Exemplo: uso prolongado de agonista (broncodilatador) de receptores adrenérgicos beta 2. Em relação aos anticoncepcionais não ocorre esta dessensibilização, a troca do anticoncepcional está associoda a outros fatores, como menor toxicidade por exemplo.
thumb|left|ligação=
Através da imagem, pode-se perceber que há uma redução da resposta farmacológica devido a dessensibilização pelo uso crônico de um medicamento e com a retirada da fármaco por um longo período de tempo (por volta de 4 meses), tem-se a resposta desejada com o retorno da administração. É importante lembrar que a dessensibilização permanece com retirada a curto prazo (uma semana).
A hiporregulação ainda pode ser classificada como homóloga, que interfere somente na resposta do fármaco que atua nos receptores estimulados. Ou pode ser heteróloga, que não interfere somente no receptor alvo, pois o fármaco (não específico) atua em outros receptores ou faz parte de uma via comum a muitos receptores. Um exemplo são os antidepressivos tricíclicos, que acabam hiporregulando diversos receptores. No caso de uma repercussão numa via comum podemos citar dessensibilização da proteína GS (comum para os receptores tipo 2). Sendo assim, qualquer fármaco/substância que dependa da via de transdução de sinal da proteína GS terá resposta reduzida.
Quanto a supersensibilidade:[]
Supersensibilidade irá ocorrer em resposta a um antagonista administrado a longo prazo que teve interrompimento abrupto da terapêutica. Exemplo: antagonista de receptor beta adrenérgico => antihipertensivo (propanolol). A utilização contínua do fármaco faz com que o receptor fique suprarregulado ou inibido. Uma vez que o medicamento é um antagonista, liga-se no sitio ativo e impede, por competição, a ligação de noradrenalina no receptor. No momento em que o paciente para subitamente de tomar o medicamento por conta própria ocorre a supersensibilização do receptor. Já que não há mais antagonista competindo, o receptor fica hipersensível a noradrenalina, o que pode gerar uma hipertensão rebote.
Caso o médico queira trocar a classe do medicamento ou acredita que o indivíduo já está com sua pressão controlada, o antihipertensivo deve ser retirado aos poucos. Retirada deve ser gradual, num período de duas semanas aproximadamente.
Ações Independentes de Receptores:[]
Alguns fármacos, através de reações bioquímicas, não necessitam de receptores para desencadear uma resposta. Esta é gerada pela ligação dos fármacos a moléculas pequenas ou íons.
Por exemplo, pode haver a neutralização do suco gástrico pelo uso de um antiácido. O bicarbonato, por reação bioquímica, neutrliza o ácido clorídrico do estômago e forma-se cloreto de sódio (sal) e ácido carbônico (depois se dissocia em CO2 e H2O).
Outro exemplo é o manitol, o qual é um diurético osmótico. Este fármaco é filtrado pelos glomérulos, mas não é reabsorvido (aumenta a osmolaridade no lúmen). Deste modo, por permanecer no lúmen tubular provoca o influxo de líquidos, portanto, favorece a diurese osmótica.
A resina de colestiramina tem uma ação sequestradora de ácidos biliares. Tem como função diminuir os níveis de colesterol. Esse fármaco se liga aos ácidos biliares e forma um complexo grande que não consegue ser absorvido pelo trato gastrointestinal e, então, é eliminado pelas fezes. Sendo assim, há menor quantidade de ácido biliar e, consequentemente, sal biliar para a digestão de lipídeos, como resultado há menor absorção de colesterol.
Quantificação da Interação Fármaco-Receptor[]

{kind=link}
Curva de interação fármaco-receptor. Farmacologia, Rang and Dale - 7ª edição.
Curva dose-resposta: Leva em consideração a concentração do fármaco utilizada e a resposta obtida para o determinado medicamento.
A resposta do fármaco aumenta conforme ele se liga aos receptores. Chega um ponto em que o fármaco atinge sua resposta máxima, neste momento já não há mais receptores livres para o fármaco se ligar (considerando um agonista total). CE50 é a concentração eficaz do fármaco para se ter 50% da resposta desejada.
Potência e Eficácia Relativa
Depende da ligação do fármaco ao receptor (afinidade). Além disso, também depende da produção da resposta (eficácia da alteração funcional que o fármaco causa no receptor).
Sendo assim, o fármaco se liga ao receptor pela afinidade e, depois de ligado, ele vai estimular o receptor. Esta estimulação intrínseca é a responsável pela geração de resposta.
O agonista total tem ligação máxima ao receptor, portanto, alta afinidade. Enquanto que a afinidade pela conformação ativa de um agonista parcial é menor, pois ele se liga tanto pela conformação ativa quanto pela conformação em repouso. Sendo assim, mesmo que se aumente a concentração de um agonista parcial para valores muito elevados nunca se alcançará um resposta máxima.
===Quantificação do Agonismo ===
A potência de um medicamento depende dos seguintes fatores: da densidade dos receptores, bem como o número de receptores em um tecido; da eficácia de um mecanismo estímulo-resposta para cada tecido (pode mudar de um lugar para outro, pois representa a atuação do fármaco para cada tecido); e da afinidade e eficácia.
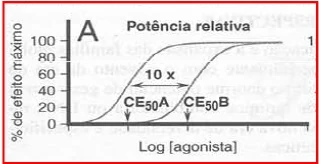
{kind=link}
Potência Relativa. Farmacologia, Rang and Dale - 7ª edição.
A figura demontra a potencia de dois agonistas de igual eficácia. Os dois fármacos tem a mesmas eficácia, pois ambos geram 100 % de resposta desejada. A grande diferença entre eles é que o fármaco A possui uma maior afinidade do que o fármaco B pelo receptor. Isto é percebido, pois é necessário uma menor concentração do fármaco A para se atingir uma resposta desejada quando comparado ao fármaco B. Além disso, o fármaco A é mais potente que o fármaco B devido esta caractarística em que necessita-se de uma menor dose para gerar resposta.
===Quantificação do Antagonismo ===
Antagonismo competitivo simples: É o antagonisma competitivo reversível. Antagonista, pois ele tem afinidade pelo receptor, mas não tem eficácia. Competitivo, pois compete com uma substância endógena do organismo uma vez que o antagonista tem afinidade pelo sítio ativo de ligação. Reversível, pois ele se liga ao receptor por uma ligação fraca/fácil de ser rompida.
Sendo assim, o antagonista é superável, em condições que há uma concentração suficientemente alta do agonista, pois a ligação é reversível. Ou seja, para um aumento da concentrção do agonista, este pode deslocar o antagonista do sítio de ligação e a substância endógena volta a se ligar.

{kind=link}
Antagonista competitivo reversível e irreversível. Farmacologia, Rang and Dale - 7ª edição.
Figura Antagonistas: A) Antagonista reversível. Mostra o desvio para a direita da curva do antagonista uma vez que há um aumento da [] do agonista. Tem-se um aumento da resposta (da substância endógeno) diminuindo o efeito inibitório do antagonista. B) Antagonista irreversível. Queda da curva/queda da resposta da substância endógena com o aumento da concentração de antagonista.
Já o que ocorre com Antagonista competitivo irreversível é diferente. Ele está ligado fortemente ao sítio ativo e não é possível deslocá-lo. Com o aumento da concentração do antagonista a resposta (pela substância endógena) vai diminuindo até chegar a zero, independetemente da concentração do agonista. Sendo assim, a antagonista irreversível não é superável. Mesmo que se aumente a concentração do agonista não haverá influência no papel do antagonista.
O Antagonista não-competitivo é um antagonista alostérico. Ele se liga a um sítio alostérico, por isso não compete com a substância endógena. Entretanto, diminui-se a afinidade do sítio ativo pelo agonista.
Figura: C) Antagonista alostérico. Com o aumento da concentração do antagonista, diminui-se a curva/resposta da substância endógena podendo chegar a zero. D) Agonista alostérico. Aumenta-se a afinidade do sítio ativo pela substância endógena. Sendo assim, a resposta desejata acontece antes do tempo normal dela (deslocamento da curva para esquerda).
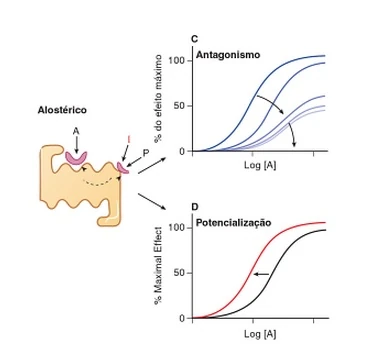
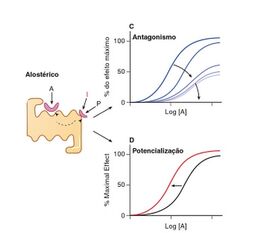
{kind=link}
Antagonista e agonista alostéricos. As Bases Farmacológicas da Terapêutica. Goodman & Gilman.
Links Externos:[]
1. Agonista e Antagonista - Quantificação
2. Receptores, agonistas e antagonistas (afinidade x eficácia)
3.Mannitol Diuretics - exemplo para ação independente de reptores
Referências:[]
- RANG, H. P.; DALE, M. M. Rang & Dale. Farmacologia. 7. ed. Rio de Janeiro: Elsevier, 2011.
- CARVALHO, GM. Anotação aula da Disciplina de Farmacologia. UNIVILLE. 27/03/2013
- GOODMAN, L.S; GILMAN, A. (eds.). As Bases Farmacológicas da Terapêutica. 12. Ed. Porto Alegre: Editora McGraw Hill, 2012.