Editor: Giovani Meneguzzi de Carvalho
Colaboradores: Amanda Lewandowski da Silva, Mariana Ribeiro, Lisiane Martins, João Francisco Petry e Pâmella Kreling
Metabolismo de Primeira Passagem
Refere-se ao metabolismo que a droga sofre antes de atingir a circulação sistêmica. A passagem do fármaco pelo organismo o degrada devido a ação de enzimas estomacais e intestinais, além de sua passagem pelo fígado. Deste modo a disponibilidade do fármaco diminui e a fração que chega na circulação sistêmica intacta é dita como biodisponibilidade.
Farmacocinética
É a resposta do organismo ao fármaco. Isto reflete as alterações na concentração do fármaco em relação à dose administrada. Os parâmetros que influenciam na disposição do medicamento são: Depuração, Volume de distribuição, Meia vida de eliminação e Biodisponibilidade.
Depuração:
Um sinônimo pode ser remoção. É a quantidade de fármaco (em volume de plasma) que está sendo eliminada do organismo por unidade de tempo. Assim, a depuração relaciona a velocidade de eliminação e a concentração plasmática do fármaco.
Estado de equilíbrio é quando a taxa de eliminação é igual a taxa administração de um fármaco, desta forma a biodisponibilidade é total. Atinge-se 94% (um valor que já é satisfatório) desse estado após quatro meias vidas (t1/2) durante a administração continua do medicamento. Durante a primeira administração se tem 100% de concentração do fármaco, depois de sofrida a meia vida, elimina-se 50%. Na segunda dose haverá 150% de concentração (100 da dose administrada e 50 da dose restante), após ocorrida a meia vida haverá 75% de concentração do fármaco no organismo. Após a quarta administração já existirá 100% de concentração do medicamento no organismo e mais 100% da dose, ocorrerá a meia vida e 100% de concentração é eliminada restando 100%.
| Dose | Quantidade de Fármaco no Organismo | Quantidade de Fármaco Eliminado (meia-vida) |
|---|---|---|
| 1 | 100% | 50% |
| 2 | 150% (100 dasegunda dose e 50 da primeira) | 75% |
| 3 | 175% | 87,5% |
| 4 | 187,5% | 93,75% |
Volume de Distribuição:
“É o volume de líquido que seria necessário para conter todo o fármaco presente no corpo na mesma concentração dosada no plasma.” Ele é diretamente proporcional a concentração de fármaco no corpo e inversamente proporcional a concentração de fármaco no sangue.
Os principais compartimentos para os quais os fármacos se distribuem são: plasma (5%), líquido intersticial (16%), líquido intracelular (35%), líquido transcelular (2%) e gordura (20%). Quando um fármaco não é lipossolúvel (polar) ele fica confinado no plasma ou líquido intersticial, justamente por não conseguir atravessar as paredes dos capilares. Já os fármacos lipossolúveis (apolares) chegam a todos os compartimentos e podem ser acumulados no tecido adiposo.
Deste modo, conclui-se que quanto maior a solubilidade de um fármaco maior será o seu volume de distribuição. E para fármacos que se ligam muito a proteínas plasmáticas, menor será o volume de distribuição, uma vez que estarão “aprisionadas” no sangue e devido à ligação não atravessarão a parede do vaso.
O volume de distribuição depende: da ligação do fármaco aos locais receptores; dos níveis de proteínas plasmáticas e teciduais (quanto menor este nível, maior será o transporte de fármaco livre); coeficiente de distribuição do tecido adiposo (quanto maior este coeficiente maior será o volume de distribuição); acúmulo em tecidos pouco irrigados (quanto menor o acúmulo maior o volume de distribuição). Além disso, o volume de distribuição pode variar com a idade, composição corporal e doenças. Por exemplo, devido a perda de albumina por cirrose, assim o fármaco fica mais livre, agindo por mais tempo.
Exemplo dado em sala: DIGOXINA
É um fármaco utilizado na insuficiência cardíaca e atua aumentando a força de contração. Por ser altamente lipossolúvel, o fármaco se distribui muito para músculos, tecido adiposo e Na+K+ - ATPase, já sua quantidade no plasma é pequena.
Varfarina e Cloroquina:
Outros exemplos dados em sala: VARFARINA é um anticoagulante oral e tem um baixo volume de distribuição. Assim, apresenta-se bastante ligado a proteínas plasmáticas e pouca quantidade nos tecidos. CLOROQUINA é um fármaco antimalárico que quase não se liga a proteínas e, portanto, uma quantidade muito pequena é encontrada no plasma. Como seu volume de distribuição é alto e seu transporte também é alto, o fármaco se acumula no tecido (retina).
Meia Vida (T1/2):
É o tempo necessário para a concentração do fármaco cair 50 %. Normalmente, é o tempo de administração entre as doses de um medicamento (de oito em oito horas; quatro em quatro).
Para um paciente com insuficiência renal crônica a depuração está diminuída, sendo assim a concentração do fármaco permanece elevada por mais tempo. Tempo da meia vida é maior.
Para um fármaco como o DIAZEPAN, medicamento tranquilizante e ansiolítico, lipofílico, que se acumula no tecido adiposo e que tem um elevado volume de distribuição. Tempo da meia vida é maior, já que a longo prazo há um aumento de sua concentração no plasma.
Portanto, T1/2 é diretamente proporcional ao volume de distribuição e inversamente proporcional a depuração.
Cinética de Primeira Ordem:
É quando a velocidade de desaparecimento de um fármaco é exponencial. Uma fração constante é eliminada por uma unidade de tempo, ou seja, 50% são sempre eliminados. Este tipo de eliminação depende da concentração plasmática, pois com o aumento da administração do medicamento, mais fármaco será eliminado. Continua-se eliminando 50%, mas este 50% representa um valor maior.
Cinética de Ordem Zero ou de Saturação:
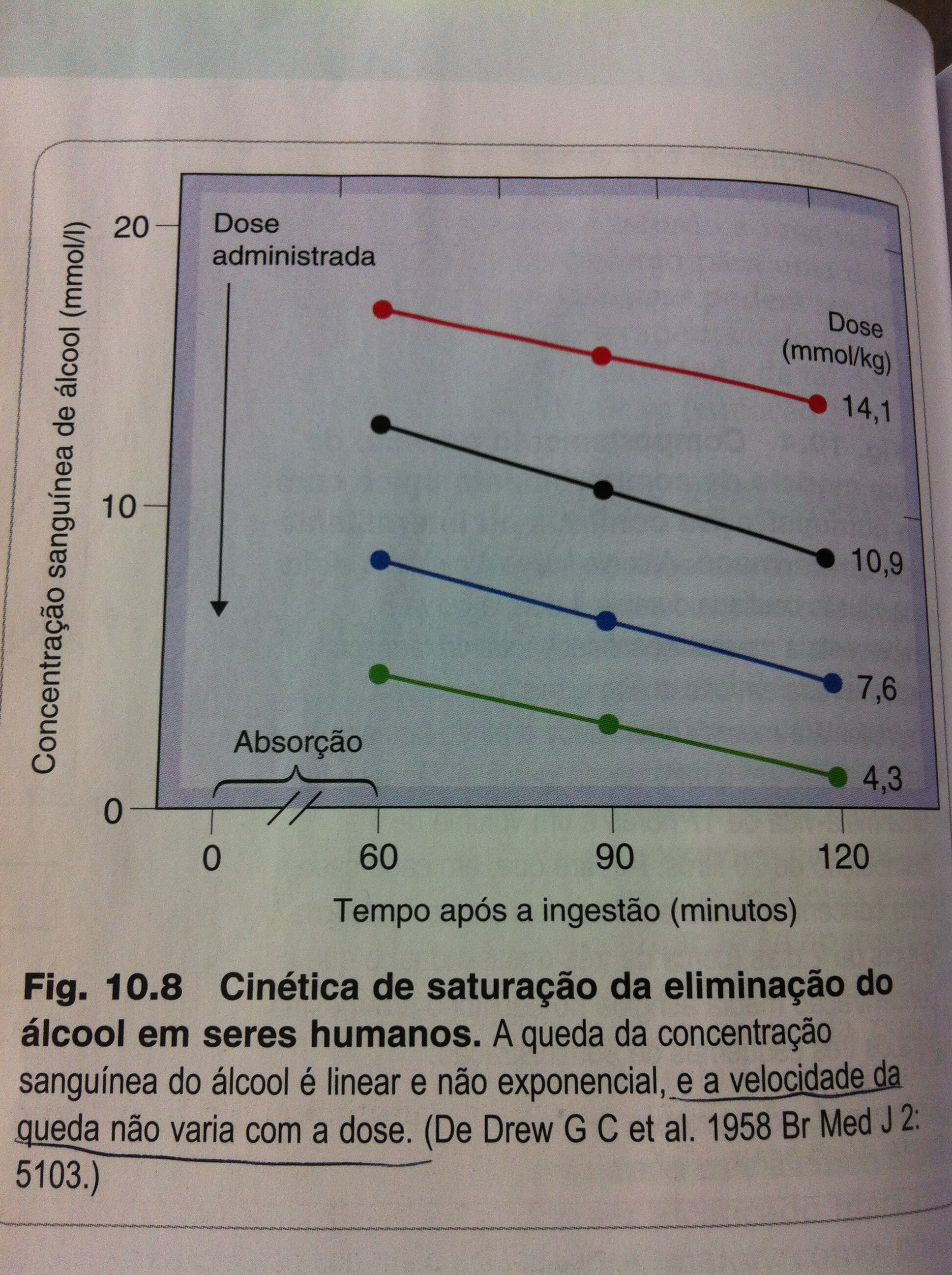
{kind=link}
Cinética de ordem zero. Fonte: RANG, H. P.; DALE, M. M. Rang & Dale. Farmacologia. 7. ed. Rio de Janeiro: Elsevier, 2011.
Este tipo de cinética difere da anterior, pois não segue um padrão exponencial de eliminação. Inicialmente é linear, pois o fármaco é removido a uma quantidade fixa, a qual não depende da concentração plasmática. Ou seja, se a dose aumentar não aumenta a eliminação. Isto ocorre com o etanol, a fenitoína (anticonvulsivante) e o salicilato.
Pode-se explicar a cinética de ordem 0 pela saturação da enzima que metaboliza a substancia. Por exemplo, no caso do etanol, a enzima álcool desidrogenase atinge seu funcionamento máximo/velocidade de oxidação em baixas concentrações do etanol. Se a concentração ingerida for maior que a oxidada haverá acúmulo de álcool no sangue. Assim, a dose tem um efeito muito importante na duração da ação do medicamento já que uma quantidade constante é eliminada.
Fatores importantes a serem considerados para a cinética de saturação:
Neste caso, portanto, a concentração plasmática no estado de equilíbrio é diretamente proporcional a dose.
- A velocidade de queda da concentração plasmática do fármaco não depende da dose.
- A interrupção da administração leva a eliminação lenta até chegar à depuração de primeira ordem, momento em que a enzima não está mais saturada.
Modelo de Compartimento Único

{kind=link}
Admite que o ser humano compreende um único compartimento.
Na realidade, este modelo é equivocado e não é aceito.Afirma-se que toda a administração do fármaco ocorre diretamente no compartimento central e que a distribuição do fármaco é instantânea por todo o volume. Isto é errôneo, pois o fluxo de sangue não é igual para todos os tecidos.
Modelo de Múltiplos Compartimentos
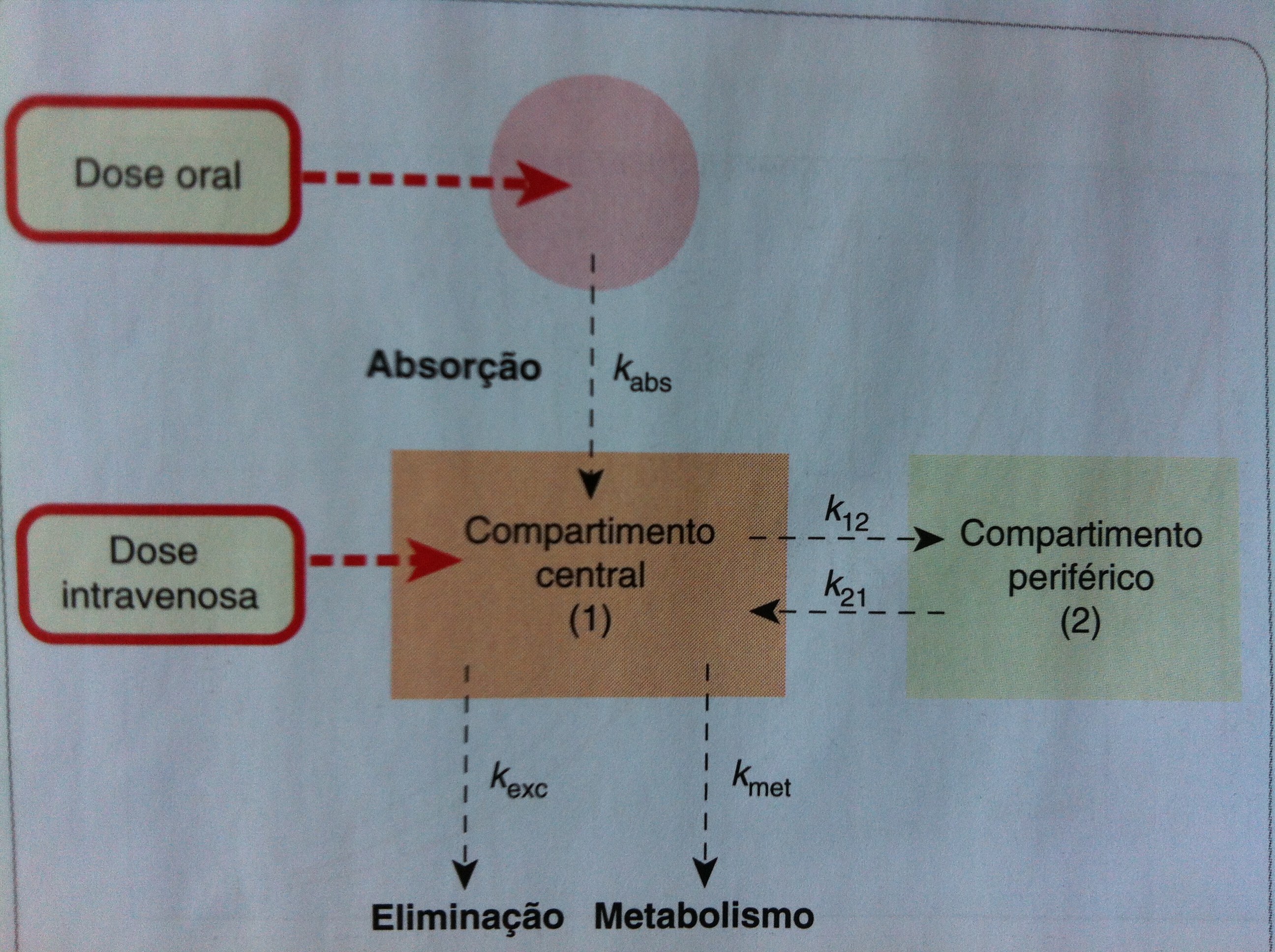
{kind=link}
Compartimentos Múltiplos. Fonte: RANG, H. P.; DALE, M. M. Rang & Dale. Farmacologia. 7. ed. Rio de Janeiro: Elsevier, 2011.
É o modelo que mais se aproxima da realidade. Divide-se em dois compartimentos: um compartimento central, em que estão o sangue e órgãos altamente irrigados (coração, cérebro, pulmões, rins e fígado); e um compartimento periférico/final/tecidual, em que estão os tecidos menos irrigados (músculo, pele, tecido adiposo e ossos). Para uma administração intra-venosa, o fármaco se distribui primeiro para o compartimento central e posteriormente para os compartimentos finais.
Um fator importante a ser considerado neste modelo é que as moléculas do fármaco só podem entrar ou sair do compartimento tecidual por meio do compartimento central.
Em uma alteração do fluxo sanguíneo como, por exemplo, na cirrose hepática, pode haver um acúmulo de fármaco em outros tecidos já que o fluxo de sangue no fígado está prejudicado.
Em uma hipoperfusão tecidual (redução do fluxo sanguíneo), o compartimento final vai receber menos fármaco e haverá uma maior concentração de fármaco no compartimento central, pois ele é mais irrigado.
Taxa de Absorção
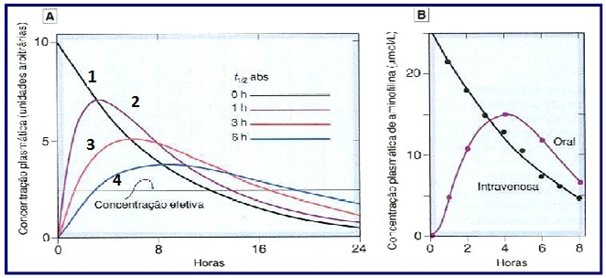
{kind=link}
Efeito da absorção lenta de um fármaco sobre sua concentração plasmática. Fonte: RANG, H. P.; DALE, M. M. Rang & Dale. Farmacologia. 7. ed. Rio de Janeiro: Elsevier, 2011.
Na administração intravenosa rápida, demonstrada na curva 1, o início da curva logo após a administração já está no pico de concentração, ou seja, sua concentração plasmática é muito alta e vai diminuído progressivamente. A curva 2 representa a administração por infusão lenta, em que o pico de concentração aparece um pouco mais tarde que o da curva 1 e também um pouco mais baixo. A curva 3 representa uma administração via oral, percebe-se que o pico de concentração é mais baixo e aparece mais tardiamente que o da curva 2. A curva 4 representa uma preparação de concentração controlada.
Através da análise do gráfico percebe-se que quanto mais lenta é a absorção, o pico de concentração aparece mais tarde e esta concentração é, também, mais baixa com um pico menos acentuado.
Links Externos:
Clearance of Drugs - depuração de fármacos junto com uma visão matemática.
Comparação entre os tipos de Cinética - linear e não linear
Referências Bibliográficas:
- GOODMAN, L.S; GILMAN, A. (eds.). As Bases Farmacológicas da Terapêutica. 12. Ed. Porto Alegre: Editora McGraw Hill, 2012.
- Giovani Meneguzzi de Carvalho. Anotação aula da Disciplina de Farmacologia. UNIVILLE. 27/03/2013
- RANG, H. P.; DALE, M. M. Rang & Dale. Farmacologia. 7. ed. Rio de Janeiro: Elsevier, 2011.